ڈVضف ،¤ ±±¾© ،¤ةد؛£ ،¤ ةîغع
yuanxing@yuanxingmed.com
 020-29820-234
020-29820-234
020-29820-234
020-29820-234
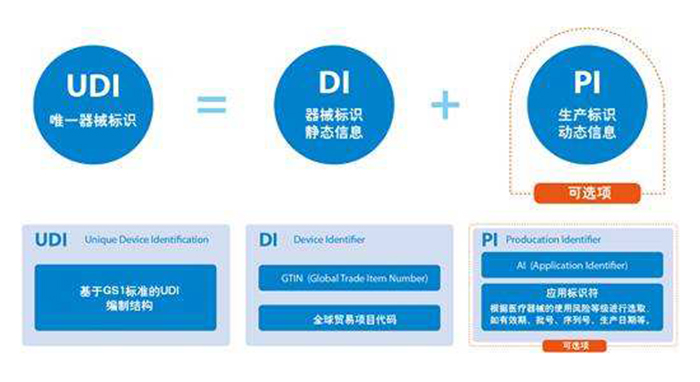
| “د¤£¬½üبصµؤڑWضق×h•صت½ح¨ك^ƒةي—·¨زژ²ف°¸£¬Œ¦لt¯ںشO‚ن¼°َwحâش\”àلt¯ںشO‚نŒچت©¸ü‡ہ¸ٌµؤ°²ب«زژ¶¨£¬´ثي—زژ¶¨Œ¢Œ¦ؤ؟ا°µؤ±O¹ـضئ¶بژ§پيضط´َ¸ؤ׃،£ذآ·¨زژµؤشِذق£¬ف^ك^ب¥µؤض¸ءîض÷زھسذزشدآژ×üc¸ؤ׃£؛ ،،،،ز»تالt¯ںئ÷²ؤµؤ¶¨ءxإc·ض¼‰£¬”U´َئن؛ةw·¶‡ْدآµؤلt²ؤإcض÷„ست½ض²بëلt²ؤ£¬Œ¢·الt¯ںسأح¾لt²ؤ¼{بëئنضذ،£ ،،،،¶تاَwحâش\”àشO‚ن·ضîگµؤ¸üذآ£¬زئ³ءثسذêPَwحâش\”à،¢ثژسأ®aئ·،¢»¯ٹyئ·،¢ت³ئ·،¢؛¬سذبث»ٍ„سخï¼ڑ°û»ٍ»î½M؟—ض®®aئ·¼{بëلt²ؤµؤ·¶‡ْ،£ ،،،،بتاشِڈٹإR´²شu¹ہإcإR´²×C“±O¹ـ£¬زھاَلt²ؤضئشىةج±طيڑكMذذإR´²ذشؤـرذ¾؟£¬زہئ÷²ؤïLëUµب¼‰جل¹©ئن°²ب«ذش¼°ذشؤـدà·QµؤإR´²×C“£¬²¢تص¼¯،¢±£ءôةدتذ؛َµؤإR´²ظYءد£¬زش³ضہmشu¹ہ®aئ·µؤ“شع°²ب«ïLëU،£ ،،،،ثؤتاجلة®aئ·Œ¦»¼صكµؤح¸أ÷¶ب؛ح؟ة×·ثفذش£¬أ÷´_زژ·¶لt²ؤةدتذا°³¾ك‚نCEصJ×Cح⣬ز²‘ھدبشع®aئ·کث؛ةدکثت¾خ¨ز»ئ÷²ؤکث×R£¨UDI£©£¬²إشتشSشعڑWأث·¶‡ْƒبنNتغ،£UDIµؤت¹سأ؟ةجل¸كضئشىةج؛حسذêP®”¾ض؟ة×·ثفدàêPئ÷²ؤ¹©‘ھوœµؤؤـء¦£¬ح¬•r´ظت¹´وشع°²ب«ïLëUµؤئ÷²ؤµأµ½¼´•r،¢¸كذ§آتµؤصظ»ط،£ ،،،،خهتاةدتذ؛َ±O¶½دµ½y£¬°üہ¨ةدتذ؛َإR´²×·غ™ض¸ؤد£¬زش¸üذآµؤلt¯ںئ÷ذµµؤإR´²شu¹ہ،¢²»ء¼·´‘ھتآ¼¼°°²ب«دàêPظYءد£¬²¢²»شظزشةجکI™Cأـ±£×o،£ ،،،،“ءث½â£¬لt¯ںشO‚نذآ·¨زژŒ¢شعزژہسعڑWأث،¶¹ظ·½¹«ˆَ،·ةد؟¯³ِµؤ3ؤê؛َé_ت¼ةْذ§£¬¶َّwحâش\”àلt¯ںشO‚ن„tشع5ؤê؛َةْذ§،£زٍ´ث£¬‡ّƒب³ِ؟عةج؟ة½èضْك@¶خ•régپيكm‘ھذآزژ£¬¼°•r¸ْكMدàêPذإد¢£¬×ِ؛أ®aئ·ص{صû¹¤×÷،£ ،،،،¾‰إdلt¯ںŒ£کIضآء¦سعلt¯ںئ÷ذµ®aئ·×¢ƒشإc‚ن°¸،¢لt¯ںئ÷ذµةْ®aشS؟ة×C،¢لt¯ںئ÷ذµ½› IشS؟ة×C،¢ز»îگلt¯ںئ÷ذµةْ®a‚ن°¸،¢¶îگلt¯ںئ÷ذµ½› I‚ن°¸،¢لt¯ںئ÷ذµإR´²ش‡ٍ،¢CEصJ×C،¢FDA×¢ƒش،¢ISO13485ظ|ء؟¹ـہيَwدµصJ×C،¢GMPصJ×Cµب،£ |
0 —lشuƒr